本文來自:平安研究,作者:葉寅 黃施齊
摘要
小核酸藥物相比現有療法優勢顯著。小核酸藥物相比現有的小分子和抗體藥物具有靶點篩選快、研發成功率高、不易產生耐藥性、更廣治療領域和長效性等優點,具有較大發展潛力。從數據來看,目前小核酸藥物已在臨牀中初步展現出治癒疾病、替代現有療法和填補空白適應症的能力,相繼出現Leqvio和Spinraza等重磅品種。未來伴隨技術持續進步,小核酸藥物有望成爲繼小分子和抗體之後具有顛覆性的新主流療法。
市場潛力巨大,但發展仍處於早期階段。由於小核酸藥物有望涵蓋更豐富的適應症,因此其潛在市場規模十分廣闊,預計到2025年全球小核酸藥物銷售額將突破100億美元。截至目前,全球已有14款小核酸藥物上市,此外還有279款藥物在研,其中60%仍處於臨牀前階段。而我國由於起步較晚,因此目前僅16款產品進入臨牀,與全球相比差距較大,其中由中國團隊自主開發、進入Ⅱ期的產品數更少。考慮到我國患者需求較大,因此隨着研發推進,我國小核酸行業仍有較大發展空間。
技術難關有待進一步突破。雖然在研小核酸數量較多,但多數藥物的靶器官集中於肝臟或局部給藥,主要是由於其他組織的特異性遞送系統仍有待進一步開發。目前全球小核酸藥物平均每年獲批約2款,佔FDA批準藥物總數的5%左右,其處境類似20年前的抗體藥物。與單抗類似,小核酸藥物也屬於技術驅動型行業,參考Alnylam從I期到III期接近60%的成功率,我們認爲,一旦技術難關被攻破,小核酸藥物潛力將有望被全面激發,行業迎來快速發展期。
投資建議:小核酸藥物已在臨牀上展現顯著治療潛力,市場具有較大發展空間以及較高壁壘,在賽道中我們建議關注以下幾類企業:1)擁有自主技術平臺的企業。小核酸藥物的設計僅需靶標mRNA序列,且不需要大規模藥物篩選,因此早期藥物序列設計壁壘相對較低,其核心壁壘在於化學修飾和遞送系統的自主專利平臺建設,保障公司的持續造血能力,形成先發優勢;2)產品已進入臨牀後期的企業。與其他成熟技術不同,全球小核酸藥物目前多數仍處於早期臨牀階段,因此雖然很多企業已初步形成自主平臺,但其功能性尚未得到驗證。鑑於我國小核酸行業還在早期技術積累和專利平臺建立階段,因此如果企業已有較強臨牀數據驗證其平臺能力,能夠確保其行業領先地位,形成技術護城河。3)具備原料藥生產和研發服務能力的企業。小核酸原料藥生產需符合GMP要求,在工藝開發、放大和質量控制上存在較高壁壘,國內有能力生產原料藥及提供開發生產服務的企業目前較爲稀缺,而伴隨國內小核酸藥物研發進入快速發展期,上下遊產業鏈有望迎來爆發,建議關注相關企業。
風險提示:1)研發失敗風險;2)被新技術取代風險;3)競爭加劇風險;4)政策風險。
01
作用於蛋白質上遊的創新機制藥物
1.1 相比於靶向蛋白質的療法具有先天優勢
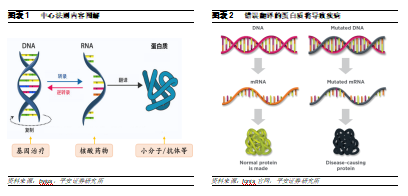
錯誤轉錄翻譯的蛋白質是導致疾病的原因之一。中心法則是Francis Crick於1957年提出的,闡明瞭遺傳信息在細胞內生物大分子間轉移的基本法則:DNA分子中的遺傳信息轉錄到RNA分子中,再由RNA翻譯生成體內各種蛋白質,蛋白質的主要功能是作爲生物體的結構成分和調節新陳代謝活動,從而維持機體正常功能。而DNA的突變或轉錄翻譯的錯誤將產生非正常功能的蛋白質,從而可能導致疾病。
靶向蛋白質的藥物存在顯著侷限性。具有由於非正常功能蛋白會通過錯誤信號傳導或影響細胞代謝等方式導致疾病,因此目前主流的小分子和大分子藥物均是通過靶向結合致病蛋白,調節其蛋白質功能,從而實現治療疾病的目的,這類靶點蛋白包括激酶、受體、抗原等。儘管部分小分子和抗體藥物已經在臨牀中獲得較好療效,並且具有易生產、給藥方便、穩定、精準等優勢,然而,這種靶向蛋白質的治療方式存在侷限性:
可成藥的蛋白質靶點選擇較少。蛋白只佔了基因組信息的極少部分,人類的基因組中,僅1.5%的序列編碼了蛋白質,其中和疾病相關的蛋白只佔10-15%,而在這些疾病相關的致病蛋白中,超過80%的蛋白質不能被目前常規的小分子及大分子藥物所靶向,屬於不可成藥蛋白,因此藥物靶點的選擇範圍較窄;
多數靶點仍然處於尚未發現的狀態。目前全球已批準的藥物僅可以與由0.05%的基因組所編碼的約700種蛋白質相互作用,仍然存在較多難以開發和覆蓋的靶點,需要多次試驗反覆驗證,尚未滿足臨牀需求;
需要考慮蛋白質三維結構,設計較複雜。小分子藥物主要通過靶向蛋白質結合口袋發揮作用,然而蛋白質並非靜態結構,在體內發揮作用時可能會發生變構,進一步加大藥物開發難度,因此早期藥物開發和篩選過程複雜;
應用範圍受到結合位點限制。抗體藥物的結合位點主要在於細胞膜表面蛋白質或細胞外,其應用受到一定限制;
需要反覆用藥甚至還會復發。這類藥物不直接調節蛋白濃度,主要調節蛋白質功能,例如抑制/促進信號傳導及催化蛋白活性等,僅起到“治標”作用,存在需要長期高頻用藥或復發的風險。
基因層面療法相比蛋白質靶向藥物具有更大潛力。如果能直接從上遊對蛋白質表達直接進行調控,將有望能夠避開以上問題,因此,實現個性化基因水平治療的藥物潛力將超過當前蛋白質靶向療法,例如核酸藥物。核酸藥物是指人工合成的具有疾病治療功能的DNA或RNA片段,能夠直接作用於致病靶基因或RNA片段,旨在改變宿主遺傳信息的編輯,具有治癒疾病的潛力,成爲“治標治本”的治療選擇。此外,由於核酸藥物理論上可以調節任何基因表達,將藥物靶點直接擴大到蛋白質上遊,因此不會受限於蛋白質的成藥性問題,有望打破不可成藥性難題。
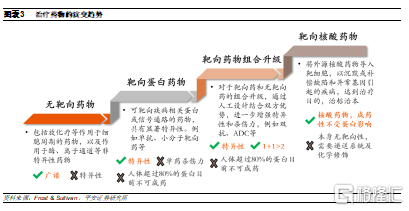
小核酸藥物是目前發展最爲成熟的基因療法之一。其中,目前已有藥品獲批上市,並且治療潛力得到驗證的核酸藥物類型主要是小核酸藥物。廣義的小核酸藥物是指長度小於30nt的寡核苷酸序列,範圍涵蓋了小幹擾核酸(siRNA)、微小RNA(miRNA)、反義核酸(ASO)和核酸適配體(Aptamer)等。相比抗體和小分子藥物,小核酸藥物具有先天優勢:
研發週期短,藥物靶點篩選快:小分子和抗體藥物需要識別某些蛋白質複雜的空間構象,因此需要大規模的藥物篩選。而小核酸藥物只需要鎖定致病基因序列,並針對該基因序列進行設計及相應RNA片段的合成,因此其早期研發速度遠遠快於其他種類藥物;
不易產生耐藥性:由於抗體和小分子主要通過調節細胞信號通路和代謝等方式發揮治療作用,因此可能會由於補償通路上調或抗原表達下降等因素產生耐藥性,而小核酸藥物直接調節上遊基因表達,因此相對不易產生耐藥性;
治療領域更廣:不受限於蛋白質的可成藥性,理論上可以設計用於靶向任何感興趣的基因,僅需要目標mRNA的序列信息,有望攻克尚無藥物的遺傳疾病和其他難治疾病;
效果持久:通常來說,小分子藥物的體內半衰期以小時計算,抗體藥物的體內半衰期以天/周計算,而由於小核酸藥物可以在體內被循環多次使用,因此能降低給藥頻次,在體內的半衰期可以按照月來計算,對很多疾病尤其是慢病的治療具有巨大的臨牀價值;
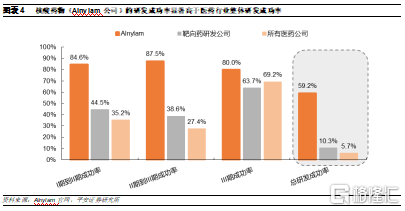
研發成功率較高:由於小核酸藥物作用機制明確,通過與mRNA完成Watson–Crick鹼基配對來實現其功能,無需契合蛋白質複雜結構,因此研發成功率相對較高。參考Alnylam公司的研發成功率,I期到III期成功率達到59.2%,相比靶向藥和整體醫藥的研發成功率高5倍。
因此,小核酸藥物的獨特優勢使其有望爲現代生物醫藥產業開闢一個全新的開發方向,進一步填補現有療法的治療空白,並且具備治癒疾病的潛力。
1.2 小核酸藥物的發展歷經機遇與挑戰
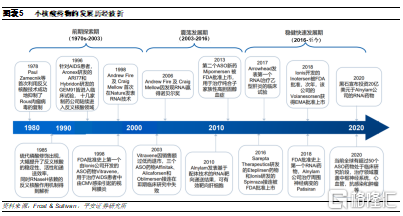
小核酸藥物作爲革新了當代藥物開發理念的創新技術,其發展過程並非一帆風順,中間歷經波折,直到近年才逐步被驗證認可,縱觀其發展歷史,大致可分爲三個階段:
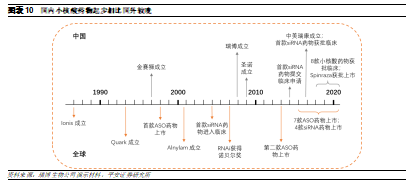
前期探索:1978年哈佛大學Zamecnik等人首次提出反義核酸概念,經過20年發展,1998年首款ASO藥物Vitravene獲批上市。同年,Andrew Fire和Craig Mello在線蟲中首次揭示了RNAi作用機制,並因此獲得諾貝爾獎。2001年Elbashir等人首次利用體外合成的siRNA實現了哺乳動物細胞中的基因表達調控,標誌着RNAi開始從研究走向臨牀應用。
震盪發展:2004年首款siRNA藥物Bevasiranib進入臨牀,小核酸藥物領域迎來蓬勃發展期,各大藥企紛紛進入該領域。然而,由於小核酸的不穩定性以及遞送效率低等缺點,多款藥物在臨牀研究中失敗,並宣佈終止開發,隨後多家大藥企放棄並出售小核酸開發平臺,行業發展一度陷入低谷期,小核酸的成藥性被質疑。
快速發展:在企業和科學家的持續研發下,GalNac遞送系統和化學修飾等技術的出現,初步解決了小核酸的遞送及穩定性問題,行業又重新迎來研發熱潮。2016-2021年多款重磅小核酸產品陸續上市,在罕見病和慢性病等領域取得重大突破,進一步驗證小核酸藥物的治療潛力。各大藥企紛紛通過自研或合作引進等方式快速佈局小核酸藥物領域,同時,資本市場也進一步助力行業和相關公司發展。
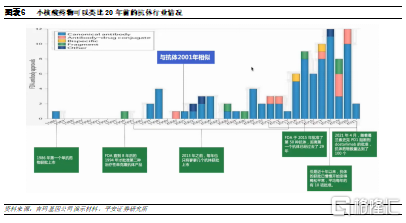
核酸藥物處境可類比20年前的抗體行業。從2016年起, 小核酸藥物每年約有2款產品獲批上市,佔FDA每年批準藥物總數的5%左右,與20年前的抗體藥物行業處境相似:在1986年首款抗體藥物上市後,到1994年纔有第二款單抗藥物上市,隨後又經歷了幾年空歇,才進入平均每年獲批2款產品的階段,直到2014年才進入快速發展期。小核酸藥物和單抗類似,均屬於技術驅動型行業,存在較高的開發壁壘,未來隨着技術難關被逐個攻破,小核酸藥物也有望進入全面發展階段。
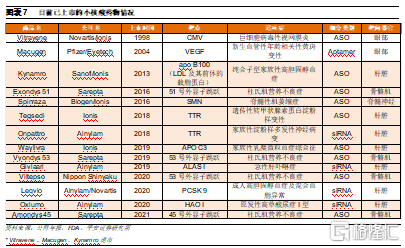
已有超過10款小核酸藥物獲批上市。截至目前,全球已獲批上市的小核酸藥物共有14款,包括4款siRNA藥物和9款ASO,1款核酸適配體,約80%的產品是2015年以後上市,其中最早上市的3款藥物目前已經退市。從適應症佈局來看,已上市小核酸藥物大部分是針對遺傳病,其中11款藥物獲得FDA或歐盟的孤兒藥認證,同時也是該疾病領域的首個藥物,一定程度上滿足了尚無治療手段的罕見患者的需求。各大企業均選擇從罕見病入手的可能原因包括:
1)遺傳罕見病靶標明確,設計上更快捷安全;2)罕見病患者少,且缺乏治療藥物,臨牀開發速度短平快;3)相比於需要長期用藥的慢性病,罕見病對於安全性的要求相對較低,適合新技術。從靶向器官來看,均屬於局部給藥或肝臟遞送給藥。
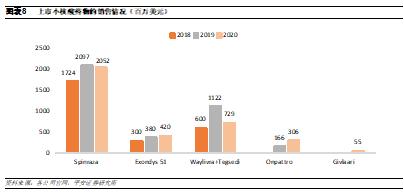
上市小核酸藥物中已出現重磅品種。2020年全球小核酸藥物銷售額在35億美元左右,其中銷售最高的是Ionis和Biogen合作開發的Spinraza,作爲全球首款獲批用於治療脊髓性肌萎縮症的藥物,其在2020年實現銷售收入20.52億美元。脊髓性肌萎縮症是一種罕見致命性遺傳病,患者主要表現爲全身肌肉萎縮無力,身體逐漸喪失各種運動功能,甚至是呼吸和吞嚥。脊髓性肌萎縮症在新生兒中的患病率爲1:6000-1:10000,中國大約有患兒3~5萬人,Spinraza的上市爲患者提供了更多治療選擇。小核酸藥物雖然售價昂貴,但由於目前上市的產品主要集中在罕見病領域,針對的患者羣體數量較爲有限,因此小核酸藥物的銷售額還未出現爆發式增長,未來伴隨研發的持續推進,有望出現更多重磅品種。
02
小核酸行業整體具有較高景氣度
2.1 小核酸藥物的市場潛力較大
全球小核酸行業駛入發展快車道。考慮到小核酸藥物的設計和開發不會受限於蛋白質的可成藥性及靶點的發現,未來隨着遞送系統和修飾技術的持續進步,小核酸藥物有望涵蓋更廣的適應症及取代部分現有療法,潛在市場規模十分廣闊。根據灼識諮詢數據,預計到2025年全球小核酸藥物銷售額將突破100億美元,其中RNAi療法憑藉其較爲顯著的效果有望能夠實現快速增長,到2025年預計將達到45億美元,複合增速達到66%。
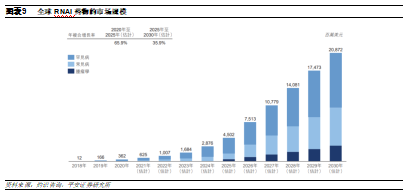
我國小核酸藥物市場仍然處於發展初期。與全球相比,我國小核酸藥物的開發起步較晚,第一家小核酸藥物研發企業的成立時間相比Ionis晚10年,首款siRNA藥物的臨牀獲批時間相比全球晚了11年。因此目前國內的小核酸行業仍然處於發展初期階段,僅一款產品獲批上市,爲Ionis 的Spinraza。但由於國內患者羣體基數較大、需求較多,因此未來伴隨小核酸藥物開發的持續推進,以及國內企業的技術逐步成熟,我國小核酸藥物市場有望迎來快速發展。
2.2 國內外小核酸研發進展差距較大
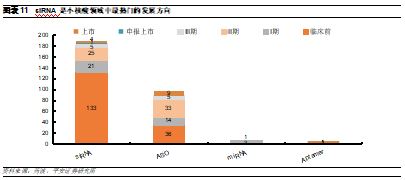
ASO和siRNA目前是小核酸藥物領域的熱點賽道。根據藥渡數據庫的不完全統計,目前全球上市和在研的小核酸藥物共有293款,其中ASO作爲最早發展的藥物類型,目前上市藥物數量最多。siRNA雖然起步相比ASO較晚,但憑藉其更高的效率和長效性吸引了更多的企業,目前共有185款在研產品,佔全部研發管線比例達到65%,已成爲主流RNAi藥物,但多數仍處於臨牀前階段。而miRNA和Aptamer的開發技術仍然有待進一步發展,目前在研藥物數量較少。
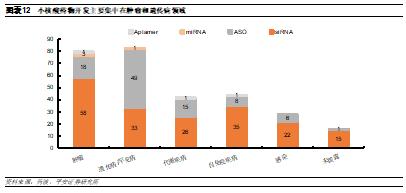
遺傳病和腫瘤是在研小核酸藥物的主要適應症。小核酸藥物的適應症涵蓋範圍較廣,包括腫瘤、罕見病、代謝疾病、自免疫疾病以及感染等,從管線數量來看,目前遺傳病和腫瘤是最爲熱門的開發領域。其中,遺傳病多數仍然沒有成熟的治療方案,因此整體競爭格局相對較好,有望爲患者帶來治療希望。從不同藥物類型來看,目前siRNA藥物多數集中在腫瘤領域,而ASO藥物多數集中在遺傳病領域,這可能是由於siRNA僅能敲低基因表達因此在遺傳病中作用受限所致。
我國小核酸藥物行業發展和國外相比差距較大。由於我國小核酸藥物開發起步較晚,因此目前獲批進入臨牀試驗的項目數量與全球相比差距較大,僅16款產品處於臨牀階段,其中多數來自於國外企業,而由中國團隊自主開發、進入Ⅱ期的產品數目較少。此外,從產品類型和靶點來看,目前我國小核酸藥物開發仍然處於跟隨和模仿階段,多數企業選擇的是國外已上市藥物的me-too類產品。未來伴隨我國小核酸藥物開發企業的研發能力提升,有望逐步進入差異化創新和突破型創新階段。
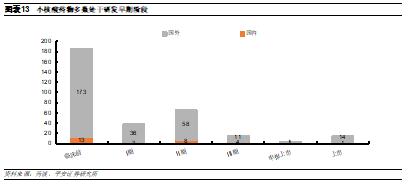
我國小核酸藥物的適應症佈局和國外具有差異性。從適應症佈局情況來看,國外在研產品數量最多的適應症爲遺傳病、腫瘤和自免,而我國在研小核酸藥物主要集中在國內的大疾病賽道,包括感染、腫瘤和代謝疾病等。
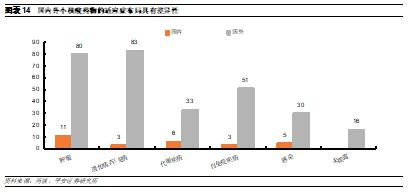
2.3 行業內領軍者多爲新興企業
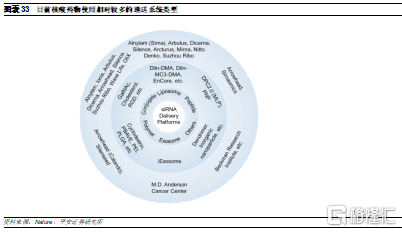
全球領先的少數小核酸廠家均爲中小型Biotech公司。由於ASO和siRNA的開發技術區別較大,因此小核酸藥物開發企業主要分爲兩類:1)專注於ASO藥物技術的企業,例如Ionis和Sarepta。其中Ionis是最早進入小核酸領域的企業,目前研發管線數量也最多,而Sarepta主攻杜氏肌營養不良症,已經形成了豐富的產品梯隊;2)專注於siRNA的企業,例如Alnylam和Arrowhead等。其中Alnylam於自2018年迎來密集收穫期,成爲首家實現商業化的siRNA領軍企業,目前已有4款上市藥物。而根據藥渡數據庫,目前瑞博生物和聖諾製藥是國內擁有小核酸藥物管線最多的企業。
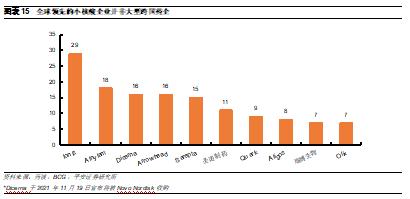
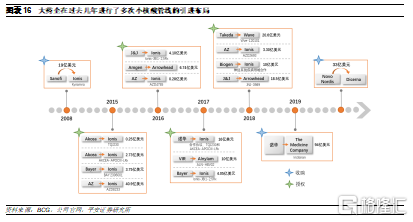
大藥企目前主要通過收購或引進等方式佈局小核酸領域。回顧小核酸藥物發展歷史,由於行業一度陷入過低谷期,彼時多家大藥企紛紛出售或終止小核酸管線,因此小核酸藥物技術進步主要是由這些中小型Biotech公司推動,從而形成了目前的企業競爭格局。而在技術難關被逐步攻破後,爲重新進入小核酸賽道,加快管線佈局,很多大型藥企在過去5年紛紛與這類Biotech公司達成引進和合作開發協議,例如Roche、AZ、J&J和諾華等,行業整體景氣度較高。此外,近日諾和諾德宣佈將以33億美元的收購Dicerna公司,未來小核酸行業也將逐步迎來大藥企的併購期。
因此,目前小核酸藥物行業整體具有較高景氣度、良好競爭格局、較大潛在市場規模的特性,行業具有較多機會:1)理論上可以靶向任何基因,伴隨技術持續進步,具有涵蓋所有疾病領域的潛力;2)相比現有的小分子和抗體藥物具有長效、高效和設計簡單等優勢,有望能夠取代現有療法,實現治標治本;3)具有較高技術和專利壁壘,因此行業競爭格局較好,玩家相對較少,且具有先發優勢。
03
RNAi是小核酸藥物發揮作用的重要機制之一
RNAi相比ASO作用效率更高。從目前小核酸藥物管線來看,基本上可以分爲ASO和siRNA兩大藥物類型,這兩類藥物的作用機制具有較大差別,其中siRNA藥物主要通過RNAi機制對靶基因進行調控。RNAi(RNA interfering,RNA幹擾)是一種細胞內源性調控機制,可以導致序列特異性基因沉默。與ASO相比,RNAi的優點在於可以反覆多次引導靶mRNA切割,具有更高的效率。目前已知具有RNAi作用的RNA主要分爲三類:siRNA(small interfering RNA,小幹擾RNA),micro RNA(miRNA),PIWI-interacting RNA(pi RNA)。其中,siRNA藥物由於療效較好且技術取得突破,現已成爲最受關注的一類技術。
3.1 siRNA藥物是當下最熱門的研發方向
siRNA可以在體內多次介導靶mRNA沉默作用。siRNA是與靶基因互補的長度爲21-25 nt的小片段雙鏈RNA,於1999年由英國科學家Hamilton在植物中首先發現,到2001年,Elbashir等科學家已成功合成siRNA,並發現將其轉入HELA細胞後能夠引發特異性沉默。經過研究發現,siRNA是一種降解mRNA的後轉錄基因沉默(PTGS),通過特異性誘導靶mRNA降解從而導致細胞基因靶向性沉默的現象,作用機制主要分爲幾個階段:
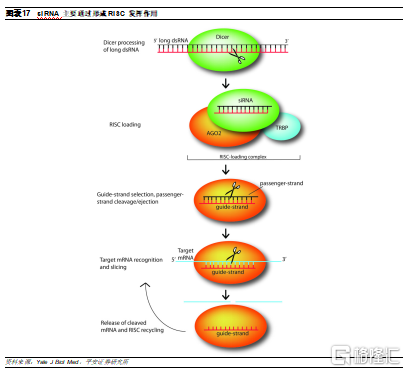
首先,外源性或內源性雙鏈RNA(dsRNA)被RNase III(例如Dicer)切割成約21-25 nt具有活性的siRNA結構。
隨後,Dicer將在RNA結合蛋白TRBP的幫助下將雙鏈siRNA加載到Argonaute(AGO2)蛋白,形成複合體(RNA-induced silencing complex,RISC)。然後,siRNA將解開雙鏈,其反義鏈與靶mRNA結合後,AGO2將特異性降解靶mRNA,從而抑制其翻譯。
最後,被切除的靶mRNA被釋放,RISC被回收,使用相同的加載引導鏈再進行幾輪切片。此外,siRNA可以在RNA聚合酶的作用下再次形成dsRNA,從而重新參與以上過程。
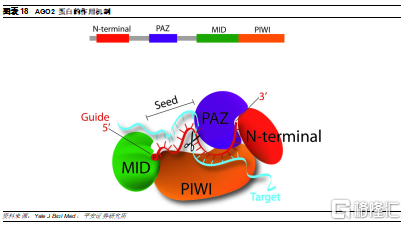
RISC中發揮沉默靶基因作用的主要部分是Argonaute蛋白(AGO2)。爲了實現siRNA介導的沉默,AGO2需要連接siRNA反義鏈,切掉隨從鏈,隨後在保持與反義鏈結合的情況下,經歷多個靶mRNA識別、切割和釋放循環。AGO2具有三個功能域,PAZ、MID和PIWI,其中PIWI結構域具有RNaseH樣的摺疊情況,是賦予其切割活性的功能域。PAZ結構域具有RNA結合作用,能夠識別單鏈RNA的3’端,起到了鉚定向導鏈的3’端的作用,5'端則插入MID和PIWI結構域之間,從而便於PIWI結構與實現切割功能。
siRNA具有獨特的成藥優勢。基於RNAi的沉默技術可用於爲多種疾病設計治療方式,主要是由一個或幾個基因引起的疾病,例如遺傳缺陷、病毒性疾病、自身免疫性疾病和癌症。鑑於其獨特的作用機制優勢,siRNA具有較大發展潛力,是目前最爲熱門的小核酸開發領域之一:
高特異性。siRNA與其靶標的結合具有高度選擇性,通過使用大約mRNA全長來識別靶序列並介導其切割。可以區分僅相差一個核苷酸的序列,這種高度結合特異性使siRNA成爲適合用於疾病治療的工具;
靶點選擇不受限制。siRNA可以沉默基因組中幾乎任何基因的表達,具有廣泛的治療潛力,有望能夠靶向現有治療方式中“無法成藥”的靶點;
治療效率高。由於單個siRNA引導鏈可以在多輪mRNA切割中循環使用,因此在正確的觸發條件下,可以實現較高治療效率。
3.2 siRNA藥物有望能夠取代現有療法,甚至實現乙肝治癒
應用方面,使用玻璃體內注射治療年齡相關性黃斑變性 (AMD) 和糖尿病性黃斑水腫 (DME) 的視力喪失是siRNA的首批臨牀應用之一,因爲這些藥物可以直接遞送至眼組織以靶向良好。從現有的臨牀數據來看,siRNA藥物已經初步展現其治療潛力:1)相比現有療法具有長效性優勢,患者依從性更好;2)具有治癒部分疾病的潛力,達到治標治本。但由於技術仍然處於發展階段,因此也存在較多失敗案例,部分開發難點有待進一步突破。
Onpattro填補空白適應症
由Alnylam開發的Onpattro於2018年上市,是首個獲得美國FDA和歐盟批準的siRNA藥物,用於治療遺傳性轉甲狀腺素蛋白介導澱粉樣變性(hATTR)。hATTR是由於TTR基因突變引起,正常情況下,TTR蛋白主要在肝臟中產生,是維生素A的載體。而TTR基因突變會導致其蛋白髮生錯誤摺疊,並且聚集成澱粉樣原纖維,積聚在多個器官中,從而導致器官和組織損傷,具體症狀包括多發性神經病和心肌病,該疾病診斷後的中位生存時間平均爲4.7年。此前較爲常用的治療方式包括肝移植、非甾體抗炎藥(Difusinal)和小分子藥(Tafamidis)。然而,這些藥物只作用於穩定TTR蛋白,對於hATTR僅有延長生存時間的效果,因此,科學家試圖直接抑制TTR的基因表達。
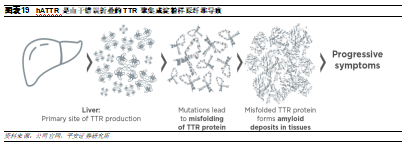
Onpattro是一種脂質納米顆粒製劑,將siRNA包裹在脂質納米顆粒(LNP)中,並通過靜脈注射直接遞送至肝臟細胞,從而沉默hATTR mRNA的表達,減少產生TTR蛋白,逐漸減少周圍神經中澱粉樣沉積物(hTTR)的積累,最終達到治療疾病的目的。數據顯示,Onpattro顯着改善了患者的生活質量和臨牀結果,56%的患者在治療18個月後表現出改善,而安慰劑治療後僅有 4% 的患者表現出改善,同時具備較好安全性,爲患者帶來了更多治療選擇。
Leqvio具有長效降脂效果,有望取代現有療法並解決耐藥問題
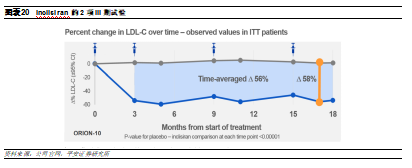
Leqvio是一款由Alnylam公司開發的用於高脂血癥的創新siRNA療法,通過將靶向PCSK9的雙鏈siRNA與GalNAc偶聯,使其特異性進入肝臟組織,從源頭上關閉PCSK9的表達,從而降低LDL-C的水平,實現降脂的治療目的,並有助於改善動脈粥樣硬化患者的預後心血管疾病(ASCVD)。根據2項關鍵III期臨牀研究ORION-10(研究高脂血癥)和ORION-11(研究ASCVD)的彙總數據顯示,在隨訪超過17個月後,超過2300名患者的LDL-C水平較基線降低了52%,其中包括了他汀不耐受患者,且安全性良好,有望能夠解決耐藥問題。
此外,Leqvio相比現有療法大幅降低了用藥頻次,初始用藥每3個月一次,此後每6個月一次(而PSCK9抗體需要每2周或每月注射一次),並且僅需皮下注射方式給藥。每年注射兩次即可有效地降低血液循環中LDL-C的水平,達到降低血脂的效果,操作起來更加簡單,因此Leqvio相比現有療法在患者依從性方面有着獨一無二的治療優勢,有望能夠進一步取代現有療法。
VIR-2218和JNJ-3989有望實現乙肝功能性治癒
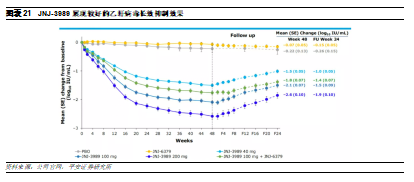
VIR-2218是一款由Vir Biotechnology開發的siRNA藥物,其採用了Alnylam的ESC+和GalNAc的遞送技術。VIR-2218通過靶向HBV X 基因的一段保守序列,可以沉默所有10種HBV基因型的所有HBV轉錄本,包括cccDNA和整合DNA,有望實現乙肝功能性治癒。在2021年歐洲肝臟大會上,公司宣佈了VIR-2218的臨牀數據,結果顯示,患者在間隔4周注射2次藥物後可以在24周內持續性的降低患者體內的HbsAg的水平。該產品與PEG-IFN-α或VIR-3434(一款HBV中和抗體)的聯用方案已進入II期臨牀。
此外,由J&J和Arrowhead共同開發的siRNA-GalNac療法JNJ-3989也展現類似治療潛力。結果顯示,患者在間隔4周注射3次藥物後,39%的患者在最後一劑給藥後的48周內展現出持久HbsAg水平下降,並具備較好耐藥性。相比現在已有的乙肝藥物只能抑制病毒複製,而不能清除病毒,siRNA藥物能夠從源頭沉默所有HBV基因產物,有望革新乙肝治療方案。
Bevasiranib和Sirna-027止步於技術難點
Bevasiranib是全球首個進入臨牀試驗的siRNA藥物,由Opko Health開發,是一種21聚體siRNA,用於治療AMD。AMD的主要發病機制是VEGF介導的脈絡膜新血管形成(CNV),因此目前AMD的主要治療方式是抑制VEGF的血管生成,包括靶向VEGF或其受體,以及抑制VEGF下遊功能。Bevasiranib則是通過下調VEGF-A 的mRNA,直接抑制VEGF的表達。而Allergan開發的siRNA藥物AGN-745(Sirna-027)則是針對VEGF受體。兩款藥物均是玻璃體內注射。
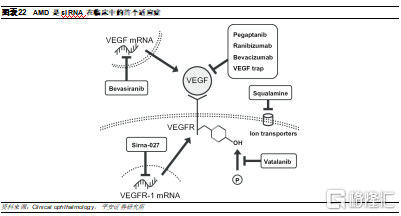
然而,這兩款藥物的臨牀均以失敗告終。Bevasiranib雖然在I期和II期臨牀中展現出生物活性,但其III期臨牀試驗由於降低視力喪失的效果不佳而終止。同樣,在完成I/II期試驗後,針對 VEGF 受體的AGN-745在II期試驗中由於脫靶效應而停止使用。因此,siRNA的首個治療嘗試由於給藥障礙以及脫靶造成的毒副作用而終止。伴隨遞送系統和修飾的持續進步,目前部分siRNA的脫靶問題已經得到初步解決。
3.3 miRNA具有較大潛力也同樣面臨技術挑戰
miRNA是內源性的短單鏈RNA分子,屬於非編碼RNA(直接作爲RNA發揮作用),與siRNA類似,miRNA也是通過RNAi機制發揮作用:
編碼miRNA的基因首先在細胞核內轉錄生成初級miRNA(pri-miRNA);
隨後,pri-miRNA將被RNase III家族酶Drosha和細胞核蛋白DGCR8的複合物切割,形成長度大約70-100nt、具有髮夾結構的前體miRNA分子(pre-miRNA);
pre-miRNA在覈輸出蛋白exportin-5的作用下被轉運到細胞質,然後被另一個RNase III家族酶Dicer和TRBP的複合物進一步切割,形成長度約19-23nt的成熟miRNA;
miRNA具有和siRNA 相同特徵的末端,隨後其引導鏈將被加載到Argonaute蛋白上以形成RISC複合體,並介導靶mRNA降解,抑制其翻譯。
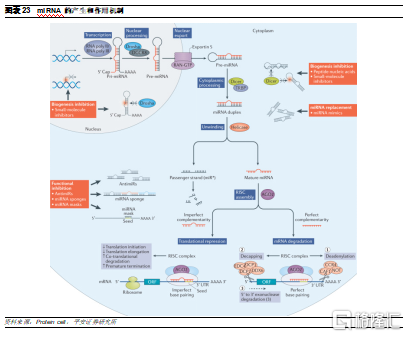
miRNA和siRNA雖然機制相同但存在差異性。雖然也是形成RISC複合體,但與siRNA需要結合靶mRNA的3’和5’端21個核苷酸不同,miRNA僅與靶mRNA的5' 端的2-8 位核苷酸結合就能發揮作用。此外,與siRNA通過AGO2發揮作用不同,miRNA通過所有4種AGO蛋白調節其靶標。雖然它們有時會像siRNA一樣導致mRNA裂解和降解,但miRNA的RISC主要通過抑制靶mRNA翻譯和去腺苷酸化降解靶mRNA來實現基因沉默。使用miRNA作爲治療具有一定優勢:
miRNA是人類細胞中天然存在的分子。與合成的化合物和ASO不同,miRNA具有處理和沉默下遊靶標的機制,人類基因組中有超過70%的部分被轉錄成非編碼RNA,選擇廣泛。
miRNA可以同時靶向多個基因。miRNA不需要以100%的互補性與其靶標完美結合,因此可以結合並抑制多種靶mRNA,從而形成一個龐大而複雜的調控網絡。與僅影響單個靶基因的siRNA或ASO相比,miRNA可能會提高治療效果,有利於解決疾病的異質性。然而,這種多基因靶向識別模式也同時增加了miRNA的脫靶概率。
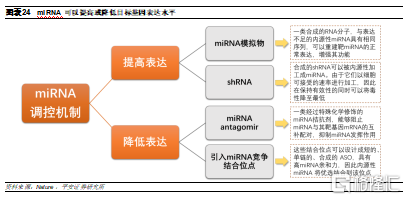
miRNA作用機制具有多樣性。與siRNA只能敲低基因表達不同,miRNA治療可以上調或下調靶miRNA表達水平,調節方式更加多樣化。
3.4 miRNA可以作爲藥物或診斷工具
miRNA的開發仍然面臨較大挑戰,因此在研管線較少。miRNA調控網絡非常複雜,其與靶mRNA的相互作用存在較多可能性,需要更準確的生物信息學預測方法,然而目前這種預測的準確率很低,僅爲26%。因此,在體外和體內驗證miRNA靶標,同時評估它們作爲藥物的適用性至關重要。體外驗證目前已經開發了許多高通量技術,然而體內動物模型仍存在侷限性,如何驗證miRNA作爲治療的適用性仍然是主要挑戰。
Remlarsen(MRG-201)能夠上調miR-29b預防纖維化
Remlarsen是由miRagen Therapeutics公司開發,是一種皮下注射的miRNA模擬藥物,旨在模擬miR-29b。miR-29b是一種細胞外基質沉積的負調節劑,可以負向調節纖維化,會在多個纖維化器官和組織中下調。臨牀數據表明,單次和多次給藥Remlarsen,可以抑制受試者皮膚傷口中纖維增生的發展,因此Remlarsen具有預防纖維化瘢痕(肥厚性瘢痕或瘢痕疙瘩)形成或預防皮膚纖維化(如硬皮病)的潛力。
Cobomarsen(MRG-106)初步展現出腫瘤治療潛力
Cobomarsen是由miRagen Therapeutics公司開發,是一種靶向miR-155的LNA antagomiR,靜脈注射。miRNA-155主要調節血液和淋巴細胞的分化和增殖,是一類在多種血液癌細胞中高度表達的致癌miRNA,特別是瀰漫性大B細胞淋巴瘤中,它被認爲是潛在的診斷和預後生物標誌物。因此,抑制 miR-155是治療血液腫瘤的重要治療策略。目前Cobomarsen在動物實驗中已經體現能夠縮小腫瘤體積的治療潛力,臨牀試驗仍在進行當中。
TTX-MC138有望能夠降低腫瘤轉移風險
TTX-MC138是由TransCode Therapeutics公司開發的、抑制MicoRNA-10b表達,miR-10b被認爲直接參與癌細胞遷移、侵襲、定植、腫瘤幹細胞和上皮間質轉化(EMT)。目前,腫瘤轉移被認爲是導致疾病惡化死亡的主要原因之一,因此,TTX-MC138通過靶向抑制miR-10b,有望能夠直接抑制癌細胞的遷移和侵襲以及腫瘤轉移,從而降低患者的腫瘤惡化和死亡風險。TTX-MC138動物試驗中展現了能夠顯著抑制多種癌症轉移的潛力,目前正在開展臨牀試驗。
MRX-34開發因脫靶毒性被終止
MRX-34是全球首個進入臨牀試驗的miRNA模擬物,由Mirna Therapeutics公司開發的脂質體納米顆粒製劑,用於模擬miR-34。miR-34是第一個被證明受抑癌基因p53直接調控的miRNA,其作爲腫瘤抑制因子,在多種癌症中檢測到被下調,因此提高其表達水平是一種重要治療策略。雖然MRX-34在針對不同癌症(包括腎細胞癌和肝細胞癌)的多項臨牀前研究中顯示出前景,然而在臨牀試驗中卻出現多起與免疫相關的嚴重不良事件,甚至出現死亡案例,因此該藥物的開發被停止。
不良事件具有免疫學特徵這一事實進一步突出了 RNA 修飾對臨牀應用的重要性,因爲此類修飾仍然是逃避 RNA 藥物免疫檢測的最重要手段之一。然而,由於 miRNA 誘導的基因調控的複雜性質,尤其是 miRNA 模擬物的化學修飾可能具有挑戰性。
miRNA 作爲診斷生物標誌物的適用性
除了具備治療潛力外,由於miRNA還是一種理想的診斷工具:1)獲取方便。miRNA 無處不在表達,可以輕鬆從體液中提取分離,並且可以通過下一代測序和逆轉錄定量PCR等技術特異性量化;2)miRNA具有特異性。其表達的變化可以指示疾病發展、進展和疾病起源組織的機制,因此量化患者miRNA變化有望可以提供更精確的診斷和治療方案定製。
例如,Quanterix和DestiNA將共同開發一種用於直接檢測和量化肝毒性生物標誌物 microRNA-122的核酸檢測方法。miR-122通常會在對乙酰氨基酚過量併發生肝損傷的患者的血清中被測量到,是一種新興的肝病生物標誌物,可提供相比現有方式更靈敏、更特異的藥物誘導肝毒性檢測。
04
ASO是目現階段發展較爲成熟的小核酸類型
4.1 ASO可以上調或下調靶mRNA表達
ASO的調節方式較爲豐富。反義寡核苷酸(antisense oligonucleotides,ASO)是合成的、與靶mRNA互補的單鏈寡核苷酸或寡核苷酸類似物,長度通常爲12-30nt。在結合靶mRNA後,ASO藥物可以通過幾種不同的方式調節其功能,包括上調和下調:
降解靶mRNA。ASO 可以設計爲內源性核酸酶(例如RNase H1或Ago2)的識別剪切位點,從而引導靶mRNA降解。其中,通過RNase H1起作用的ASO通常具有8-10個連續的 DNA 核苷酸,以支持 RNase H1 的結合和切割;而通過Ago2起作用的ASO通常以雙鏈RNA等形式傳遞到細胞中,進入細胞後,其雙鏈分子會與RISC相互作用,然後一條鏈與Ago2選擇性結合,對靶mRNA進行降解。
抑制翻譯。ASO 還可以設計靶向上遊翻譯起始密碼子AUG,阻斷RNA結合蛋白複合物(如核糖體亞基)的結合,從而抑制靶mRNA的翻譯。
剪切調控。ASO可以設計成結合內含子-外顯子連接點,可破壞剪接位點的穩定性,或置換或募集剪接因子,從而導致靶標外顯子的跳讀或內含。
增加蛋白翻譯。ASO 可以設計爲結合上遊開放閱讀框(uORF),從而增加下遊ORF翻譯的蛋白量。
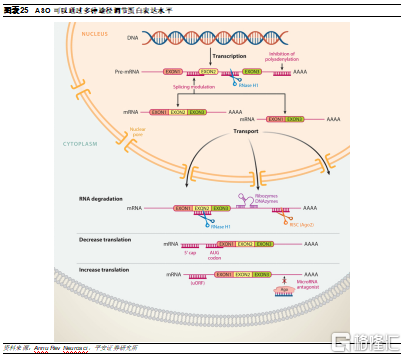
與siRNA相比,ASO具有其獨特的優勢和短板:
可以上調錶達。與siRNA僅能發揮沉默作用不同,ASO藥物還可以上調蛋白翻譯,從而治療因蛋白表達缺少而造成的疾病,例如脊髓性肌萎縮症(SMA)。
在體內的擴散效果更好。ASO作爲單鏈RNA,相比雙鏈的siRNA其親水性更弱,因此能夠更好的在組織和細胞中擴散吸收。
長效性相對較弱。然而,由於ASO是通過直接結合mRNA發揮作用,而RNAi是形成複合體在細胞內循環降解RNA,因此相比RNAi,ASO的半衰期更短,並且不具備持續性。
4.2 ASO藥物已經出現重磅品種
ASO作爲小核酸藥物領域最早發展的類型之一,目前已經有9款上市產品,佔全部上市小核酸藥物數量比例達到64%。
Spinraza憑藉較好療效成爲重磅品種
目前上市的ASO藥物中,由Ionis和Biogen 開發的Spinraza是一款明星產品,用於治療罕見病脊髓性肌萎縮症(SMA)。SMA的發生是由於運動神經元存活 (SMN) 蛋白水平低,這是由SMN1基因內的缺失或失活突變引起的。SMA 是嬰兒死亡的最常見原因之一,攜帶率爲 1:50,發病率爲萬分之一。人類基因組中存在SMN2基因,與SMN1幾乎相同,唯一的區別在於SMN2基因外顯子7的調控序列被異質核核糖蛋白(hnRNP)的結合所掩蓋,因此在翻譯時相比SMN1缺少外顯子7,會產生一種無功能且不穩定的蛋白質(SMN2Δ7)。Spinraza作爲ASO藥物,能夠與SMN2外顯子7的調控序列結合,取代hnRNP,從而在翻譯時能夠形成全長的SMN蛋白,從而增加了功能性 SMN 蛋白的水平,改善患者的運動神經元功能並減緩疾病進展。
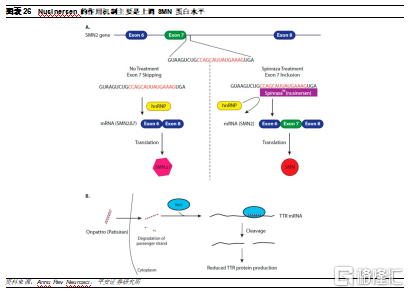
Spinraza是美國和歐洲唯一批準的SMA治療,因爲患者的運動功能得到改善、疾病進展減緩且副作用很少。作爲罕見病用藥,Spinraza定價昂貴,其一年治療費用達到75萬美元。但由於其較好的療效以及稀缺性,目前Spinraza已在全球50多個國家和地區上市,2020年銷量達到21億美元。
Vitravene是首款上市的小核酸藥物
ASO藥物是第一種廣泛用於臨牀試驗的 RNA 藥物。而首款ASO藥物Vitravene由Inois和諾華合作研發,於1998年獲FDA批準上市,治療鉅細胞病毒視網膜炎(CMV),是第一個獲批上市的反義寡核苷酸藥物。該藥物由21個硫代脫氧核苷酸組成,旨在與鉅細胞病毒的主要立早基因的mRNA互補,從而特異性抑制鉅細胞病毒關鍵蛋白的產生。臨牀數據顯示,在玻璃體注射Vitravene後,病毒複製被抑制。然而,Vitravene在市場方面並未獲得成功。伴隨抗逆轉錄病毒療法(HAART)的發展,CMV病例數急劇減少,因此,諾華分別於2002年和2006年,在歐洲和美國停止銷售該藥。
Kynamro爲遺傳病患者帶來治療選擇
在Vitravene退市後,直到2013年,ASO領域才迎來第二款上市藥物Kynamro。該款藥物由Genzyme和Isis兩家公司開發,用於治療純合性家族性高膽固醇血癥。家族性高膽固醇血癥 (FH) 是最常見和最嚴重的單基因高膽固醇血癥,其中純合子 FH最爲嚴重,患者早在兒童時期就有動脈粥樣硬化先天性心臟病,若未經治療,通常在 30 歲之前死亡。Kynamro被設計成與人類載脂蛋白 (apo) B-100特異性互補,apoB-100是一種由肝臟合成的大蛋白質,在人類脂蛋白代謝中起重要作用,是致動脈粥樣硬化脂蛋白的主要結構蛋白。Kynamro主要分佈於肝臟,通過和apoB-100形雙鏈體從而導致其被RNase H降解,從而降低血漿LDL-膽固醇和apoB-100濃度。III期臨牀數據顯示,患者在皮下注射26周Kynamro後,他們體內的低密度脂蛋白膽固醇(LDL-C)水平降低了25%,而安慰劑對照組中僅有3%的人達到這一效果。
05
核酸適配體是較爲獨特的一類小核酸藥物
5.1 核酸適配體的作用機制與其他小核酸差別較大
核酸適配體通過空間結構結合靶標。核酸適配體是合成的單鏈寡核苷酸,與其他RNA藥物通過鹼基配對發揮作用不同,當靶標存在時,核酸適配體可經過自身捲曲、摺疊形成特定的三維構型,如髮夾、凸環、四角環等,並通過範德華力、氫鍵、靜電作用、鹼基堆積力等,利用其空間結構與靶標高親和性、高特異性地結合,這一過程類似抗體-抗原的結合,因此核酸適配體又被稱爲“化學抗體”。
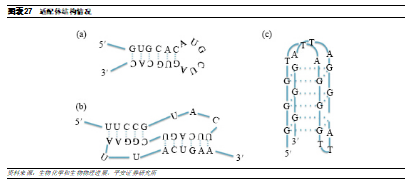
核酸適配體的選擇通常需要通過多輪體外篩選來確定其功能,這一過程通常被成爲指數富集的配體系統進化(SELEX)技術,通過SELEX可以從文庫中選擇 20-100nt的適配體,以調節類似於抗體的蛋白質功能。適配體相比其他寡核苷酸和抗體藥物具有獨特優勢:
ASO或siRNA藥物的靶標需要存在於細胞內,而適配體可以靶向細胞內、細胞外或細胞表面。
雖然核酸適配體對其目標配體的親和力和特異性可與抗體的特性相媲美,但適配體相比抗體具備成本和滲透性優勢。適配體是使用 SELEX在體外進化和鑑定的,可以被重複且經濟地大規模合成用於臨牀應用。此外,適配體的小尺寸使其組織滲透性更好。
5.2 核酸適配體目前的主要開發方向是藥物載體
適配體作爲藥物的開發熱度逐步消退
Macugen是全球首款核酸適配體藥物,由NeXstar公司研發,用於治療老年黃斑病變 (AMD)。Macugen是一個28聚體的RNA適配體,靶向血管內皮生長因子(VEGF)-165,VEGF-165是主要負責病理性眼部新血管形成和血管通透性,Macugen能夠防止VEGF-165與VEGFR結合,從而阻止血管生成以及防止VEGF-165誘導的血管通透性增加。但伴隨雷珠單抗上市,Macugen因爲療效不突出,銷售出現下滑。隨後阿柏西普等藥物的上市,使得市場競爭更激烈,這款RNA適配體藥物出局,目前已經退市。而在之後的兩款適配體藥物Fovista(抗PDGF適配體)和Reg1(抗FIXa適配體)相繼失敗後,核酸適配體作爲藥物的開發熱度逐漸消退。
適配體作爲載體是目前的主要研發方向
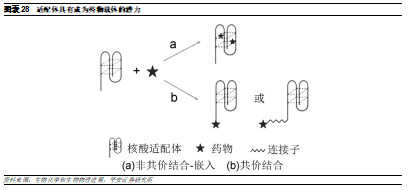
由於適配體具有良好的靶向性能,並且其三維結構有望攜帶藥物,因此其具有成爲藥物載體的潛力。例如,將阿黴素(Dox)直接結合到適配體,有望能夠進一步提高細胞毒藥物的靶向特性,降低其副作用。核酸適配體與藥物結合的方式有兩種:非共價結合(嵌入)和共價結合。
06
小核酸開發仍然面臨有待突破的技術難點
小核酸在體內發揮作用需要層層突破重圍。小核酸藥物作爲新興藥物,在面對機遇的同時也面臨着挑戰。小核酸從給藥到遞送至細胞內靶位點並發揮作用的整個途徑中面臨多重障礙:
首先需要通過血液循環到達目標組織。在細胞外,核酸藥物容易被血清和組織中的酶降解,此外,由於分子量較小,核酸藥物還容易被肝臟和腎臟快速清除,血清中裸核酸藥物的半衰期從幾分鐘到一個小時不等。同時,核酸藥物還容易被免疫系統識別並引發免疫反應。
其次需要順利到達靶細胞內。核酸藥物在通過循環達到目標組織後,需要具備血管外滲能力並被靶細胞攝取,然而裸核酸藥物的負電荷以及分子量(~12kDa)導致其不能自由通過生物膜。因此,需要解決小核酸藥物在組織和細胞中的擴散問題。
進入細胞後需要能達到靶位點。即使核酸藥物被細胞通過內吞作用攝取後,也容易被困在內吞體中,並被降解,無法順利釋放到細胞質發揮作用。體外試驗顯示,僅1%左右的核酸藥物能夠進入細胞質中,而在體內試驗則不足0.1%。因此,核酸藥物如何在靶位點積累到能夠治療的水平是一項重大挑戰。
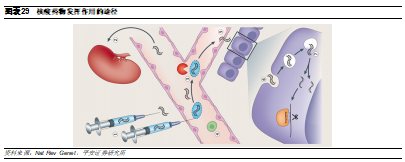
因此,爲了提高藥物遞送效率,加強治療效果,需要發明具有高效、特異性的傳遞方式,目前主要通過開發藥物載體以及對核酸藥物進行化學修飾。
6.1 化學修飾能夠提高小核酸藥物的穩定性和效果
化學修飾有利於改善小核酸藥物的缺點。未經修飾的寡核苷酸藥物成藥性並不理想,具有較多缺點,比如穩定性差易被降解、很難進入細胞、對靶標mRNA的結合親和力不佳。爲了提高治療效果,寡核苷酸藥物必須經過化學修飾,目前修飾主要包括糖、鹼基或骨架的變化。
6.1.1 骨架修飾
骨架修飾是最基本的化學修飾。連接RNA磷酸骨架的磷酸二酯鍵是核酸酶作用的化學鍵,而磷原子是核酸酶攻擊的中心,對該原子稍加改變即會大大影響酶的降解作用,因此最早被使用的就是針對骨架磷原子的化學修飾。其中使用較多的骨架修飾爲硫代磷酸,即用一個硫原子取代磷酸二酯鍵的非橋氧原子(P-S替代P-O),可以減少寡核苷酸的親水性、增加了對核酸酶降解的抵抗力以及增加了其與血漿蛋白結合,進一步增加藥物穩定性和半衰期。然而,這類化學修飾在高濃度的情況下可能會導致寡核苷酸和靶標的親和力下降,從而導致脫靶毒性和炎症反應。因此,也可以選擇硼酸磷酸鹽(P-B替代),可以將裸核酸藥物的酶抗性提高10倍以上,同時不會造成細胞毒性。
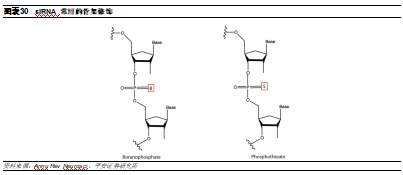
6.1.2 核糖修飾
核糖是核酸藥物最經常被修飾的部分。在一代藥物對於骨架修飾的基礎上,90年代出現了具有糖基修飾的二代寡核苷酸藥物。核糖修飾具有進一步增加靶標結合親和力、抵抗核酸酶降解、減少促炎症反應的作用,其中最重要的是對核苷酸戊糖2’-羥基(2’-OH)的修飾。2’-OH是RNA與DNA的主要區別,也是最先被酶催化並導致RNA水解的部分。因此,在該位置引入取代基後,例如甲基、氟、鹵素、胺等,能夠使siRNA具有更強的抵抗核酸酶水解的性能,增加siRNA在血清中的穩定性。但全鏈修飾將導致siRNA失去沉默活性,因此一般僅對雙鏈末端1-4個核苷酸進行修飾。此外,對正義鏈的修飾較少影響 siRNA 的活性,而反義鏈的修飾對 siRNA 基因沉默的活性影響較大。
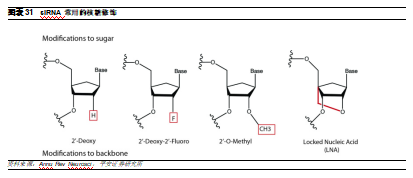
LNA是核糖修飾中最有效的一類。雖然二代寡核苷酸相比一代具有更強的親和力,但是其誘導RNase H切割靶標mRNA的效率較低,因此需要進一步提高其沉默效率。其中,目前相對有效的方式是LNA修飾。LNAs核苷酸是一類核酸類似物,其中核糖環被連接2’-O原子和4’-C原子的亞甲基橋“鎖定”,顯示出增加的熱力學穩定性和增強的核酸識別的作用。
6.1.3 鹼基修飾
鹼基修飾能夠提高小核酸藥物的沉默效果。小核酸藥物主要通過與mRNA鹼基互補形成氫鍵,從而發揮RNAi的作用,因此通過對鹼基的修飾,可以進一步加強鹼基之間的相互作用。在尿嘧啶的5位點引入溴或碘是常使用的鹼基修飾方法,如 5-溴-尿嘧啶、5-碘-尿嘧啶,可加強腺嘌呤-尿嘧啶(A-U)之間的連接,提高鹼基的相互作用,從而增強對靶 mRNA 的效應。
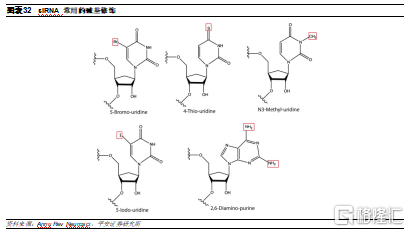
6.1.4 末端修飾
末端修飾能夠爲小核酸藥物帶來更多功能。除了修飾寡核苷酸藥物的自身結構,在其末端加上修飾可以用於調整藥代動力學特性,以及賦予siRNA雙鏈體新功能:
定點靶向細胞。通過在siRNA末端引入葉酸、肽和適體可以協助其跨細胞屏障轉運,並特異性導向部分細胞類型;
增加細胞膜穿透能力。由於siRNA帶負電荷並且具有較強親水性,因此不易與帶同樣電荷的靶細胞接觸,更不易透過由脂質雙分子層構成的細胞膜進入細胞內發揮作用。通過引入親脂性基團,如膽固醇,能加強siRNA的親脂性,增加其透過細胞膜的能力;
研究siRNA體內分佈。通過在siRNA末端附加熒光分子,可以研究其在體內的生物分佈和攝取,爲小核酸藥物的開發奠定更堅實的基礎。
6.2 遞送系統能夠使小核酸藥物被完整運輸到靶位點
小核酸藥物特性使其較難以裸RNA形式給藥。儘管化學修飾可以增強小核酸藥物的穩定性和半衰期,同時解決免疫原性的問題,但如果不能進入細胞實現胞吞,小核酸藥物依然不能發揮藥物作用。然而,作爲外源性藥物,這些基於RNA的藥物會被核酸酶降解,並且由於其分子量較大以及負電荷的特性,導致其難以穿過細胞膜發揮作用。因此,如何克服生物學障礙,實現高效跨膜和有效體內運輸以達到相應藥效一直是核酸藥物開發亟待解決的瓶頸問題,目前,遞送系統成爲提高轉送效率的關鍵辦法之一。
載體主要可以分爲病毒載體和非病毒載體:1)病毒載體效率較低且進入人體後會引起免疫反應,現在使用較少;2)非病毒載體中較爲常用的主要分爲納米顆粒和綴合物,目前被廣泛使用。
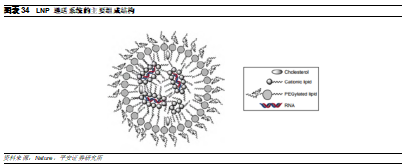
6.2.1 脂質納米粒遞送系統
LNP是應用最早的遞送系統。脂質納米粒(Lipid Nanoparticle,LNP)是使用脂質形成納米微粒,結構爲由磷脂雙層組成的囊泡。通過將核酸藥物裝載到LNP中,可保護被包裹的核酸藥物免於降解和清除,並促進其跨細胞膜運輸到目標靶位。由於成分的生物相容性和複合物的容易組裝,LNP是一種有吸引力的遞送方法,只需要混合和孵育成分,因此目前在藥物開發中使用較多。構建LNP遞送系統需要3種物質:
1)陽離子脂質,通過靜電作用包裹着核酸藥物,從而形成LNPs/siRNA複合物;
2)輔助脂質即促融合磷脂,破壞細胞膜脂質雙層結構,增加LNPs 的轉染活性;
3)聚乙二醇(PEG),通過增加膠體穩定性和保護LNPs 免受巨噬細胞的侵襲而降低免疫反應,而過多的PEG 脂質會阻礙LNPs 的細胞內化。
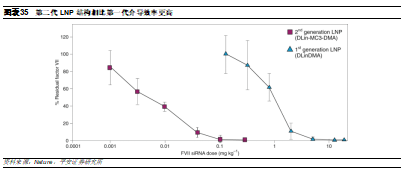
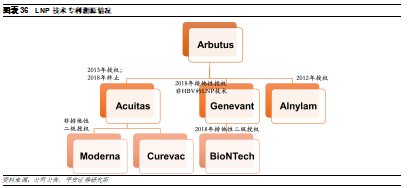
目前使用較多的亞型是可離子化LNP。最早被發現和使用的類型是陽離子LNP,然而,早期的陽離子LNP由於容易被巨噬細胞清除和產生有害的ROS等缺點,其體內應用受到很大限制。而可離子化LNP能在體內保持中性,避免被清除並降低副作用,而當進入強酸性環境如內吞體,即可以質子化形成陽離子從而與內源的陰離子脂質結合,提高跨膜效率。在Arbutus和Alnylam等公司的開發下,離子化脂質遞送系統目前已經經歷三次迭代,主要代表類型爲DLin-DMA、DLin-MC3-DMA和L319,中位有效劑量(ED50s)爲1、0.005和<0.01mg/kg。
第一代是1,2-二亞油基-3-二甲基氨基丙烷(DLinDMA),用於開發了TKM-080301、ALN-VSP和ALN-TTR0等藥物。
第二代包括DLIN-MC3-DMA,是在第一代的基礎上進一步優化,並開發了Patisiran和ALN-PCS等藥物。相比第一代,DLin-MC3-DMA具有獨特的pH依賴性電荷可變特性:酸性條件下呈正電性,而生理pH條件下呈電中性。其會在內體或溶酶體的酸性環境中質子化,導致H+和cl-離子以及水進入內體,進使得滲透腫脹破裂內體,於是siRNA可以逃離內體/溶酶體,發揮沉默作用。數據顯示,在小鼠模型中,第二代相比第一代介導基因沉默的劑量顯著降低。
由於siRNA產品需要對慢性疾病重複給藥,而二代MC3中二醇烷基尾的緩慢降解會導致重複給藥的累積和潛在毒性。爲進一步提高安全性,第三代脂質主要是增強其水解速度。例如L319採用了一種在體內容易被酯酶降解的伯酯取代烷基鏈中的兩個雙鍵之一,其代謝物爲β氧化途徑中脂肪酸的潛在基質,具有較好的安全性,並且能夠迅速通過肝臟被排泄,在肝臟中的半衰期不到一小時,因此相比第二代具有更好的耐受性和安全性,目前在BioNTech和CureVac產品中被使用。此外,通過使用其他可離子化脂類取代MC3也可以提高安全性和效價,例如Moderna開發了一類新的可電離脂質SM-102來替代MC3,提高效率。
LNP遞送系統的構建擁有較高壁壘,目前全球發展靠前的幾家企業其技術溯源也都是依靠專利授權。
此外,LNP雖然大大促進了核酸藥物的發展,但仍然有缺點,限制其使用範圍:
LNP的過敏反應較爲嚴重。在注射使用LNP遞送系統的藥物之前,患者需要使用抗組胺和激素藥物控制,因此只適用於罕見病和癌症等嚴重疾病,對於慢性病使用限制較大。
主要靶向肝臟。離子化LNPs在低PH值的環境下會和載脂蛋白E(ApoE3)相互作用,通過其介導的內吞作用富集在肝細胞中,因此由其包裹的核酸藥物也主要作用於肝臟,而對於其他器官缺乏明確的靶向性,使用範圍較爲有限。
爲解決靶向性問題,目前可以通過將靶向部分直接綴合到脂質分子上增加其對特定細胞的靶向性,使得小核酸藥物在特定組織積累,從而發揮作用。這類靶向部分可以選擇抗體或蛋白質(例如轉移素)、肽(例如,RGD或八氯丁二酯)、適配體或小分子(如透明質或葉酸),對目標組織具有高度特異性,並且在循環和組織蛋白酶中保持穩定。但這種方式會使得小核酸藥物CMC難度加大。
6.2.2 偶聯遞送系統
偶聯遞送系統已經逐步取代LNP。由於脂質分子的遞送系統仍然存在缺陷,因此各研發機構和藥企仍然持續開發新的遞送系統,近年偶聯技術在小核酸藥物中得到廣泛使用。這種綴合物偶聯的方式主要通過直接共價結合不同成分來增強小核酸藥物的靶向特異性,並降低循環中的藥物清除,這些成分主要包括脂質、肽、適體、抗體和糖(如N-乙酰半乳糖胺,GalNAc),其中最先嚐試的是脂質偶聯,而目前使用較多的主要是GalNAc偶聯。相比脂質分子,偶聯遞送系統的分子量相對更小,並且通過設計對酸敏感的連接子可以加強藥物的內含體逃逸效率。
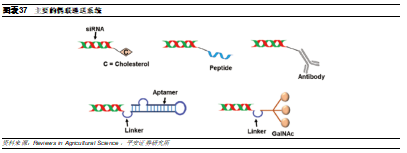
脂質偶聯
脂質偶聯存在內含體逃逸問題。脂質偶聯的RNA能夠形成類似於低密度脂蛋白(LDL)的聚合物,不但能延長循環時間,並且能與LDL受體或者其他受體結合,通過內吞進入細胞,提高遞送效率,其中較常見的是與膽固醇偶聯。由於LDL受體在肝臟中高表達,脂質偶聯藥物系統給藥也主要是靶向肝臟,但通過局部注射,也能進入皮膚,眼睛和大腦等組織。然而,最初使用偶聯遞送系統的RNA藥物在進入細胞後經常沒有功能,主要是因爲RNA分子會被“卡”在內含體內,只有約0.01%的分子能夠逃逸進入細胞質發揮功能。因此,內含體逃逸成爲偶聯給藥的限速步驟。
GalNac偶聯
GalNac已經取代LNP。GalNac是目前最常使用的偶聯遞送系統之一,已有3款基於GalNac偶聯遞送系統的藥物獲批上市。GalNAc作爲亞洛曲蛋白受體(ASGPR)的配體,是一種特異性高表達於肝細胞膜表面的內分泌受體,幾乎沒有由其他細胞表達,因此也主要用於肝臟給藥。雖然使用範圍和脂質分子一樣僅限於肝臟,但是其優勢顯著:1)安全性較好,無需類固醇預處理;2)可以進行皮下給藥,患者依從性更好;3)遞送效率較高,只需2-5mg/kg劑量即可發揮作用。由於其較爲顯著的藥效和安全性,Alnylam停止了基於LNP遞送系統的管線開發,轉而使用GalNAc。目前,Alnylam對GalNac的專利覆蓋較爲全面,涵蓋GalNac-siRNA,包括雙鏈核酸結構、限定了一些位點修飾等。Dicerna主要是通過構建單鏈結構和末尾成環等方式避開專利。
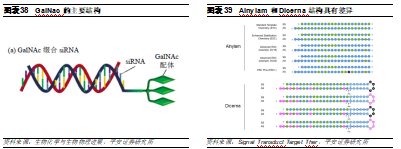
雖然Galnac特異性較強,但是其研發也並非一蹴而就。Alnylam的第一個GalNAc-siRNA藥物是Revusiran,靶向hTT基因,用於治療退行性疾病ATTR。雖然,Revusiran的II期臨牀顯示,患者在用藥12個月後血漿TTR水平降低90%,但患者死亡率的增加卻導致試驗中止。其原因可能爲該款藥物的穩定性不夠,需要大劑量給藥,因此帶來毒性。隨後,Alnylam推出了更加穩定的ESC化學骨架修飾,解決了這個問題。基於ESC修飾的第二代anti-TTR一年只需使用100mg,相比而Revusiran的28g用量,其劑量降低了280倍,療效和安全性大爲改善。除Alnylam外,近年基於GalNAc的技術平臺層出不窮,包括Dicerna的GalXC、Arrowhead的TRiM等,駛入發展快車道。
6.2.3 多聚體納米粒遞送系統
多聚體載體與LNP的遞送機制相似。多聚體載體技術在臨牀上的應用並沒有脂質體類載體廣泛,但是作爲核酸藥物的載體也表現了優異特性。鑑於其高度的化學靈活性,聚合物也是用於基於納米顆粒的遞送的常用材料。聚合物納米粒子是很有前景的基因傳遞系統,因爲它們具有穩定性和控釋性,能夠封裝大量遺傳物質,允許共同傳遞,並且可以很容易地進行表面修飾以增強穩定性、運輸特性、靶向性或攝取。目前殼聚糖、聚乳酸-乙醇酸共聚物 (PLGA)、樹枝狀大分子等納米粒子已廣泛用於遞送系統。
聚乳酸-乙醇酸共聚物 (PLGA)
LODER是一種由Silenseed公司開發的可生物降解的聚合物基質,源自聚(乳酸-共甘油)酸(PLGA),直徑爲1納米,長度爲5納米。根據Silenseed描述,LODER可以直接將RNAi藥物輸送到實體腫瘤中,並且使其緩慢而穩定地釋放。基於LODER技術,Silenseed開發了si-PT-LODER224和si-GBMT-LODER,用於治療前列腺癌和腦癌,分別處於臨牀前和研究階段。Silenseed的siG12D LODER,它封裝的siRNA針對KRAS在可植入和可降解的聚合物基質癌蛋白靶向胰腺癌
殼聚糖
殼聚糖是一種從甲殼類動物外骨骼中收穫的天然陽離子多糖。由於其生物相容性、粘膜粘附特性、免疫原性低和核酸酶抗性等特點,它是一種被廣泛研究的生物材料。在兩項獨立的研究中,優化的殼聚糖-siRNA 納米顆粒已成功地鼻內給藥以沉默小鼠肺中的 GAPDH 和 EGFP。
樹枝狀大分子
樹枝狀聚合物是高度支化的聚合物分子,可以設計成模塊化、納米尺寸的球形結構,用於siRNA遞送。主要通過兩種方式包裹核酸藥物:1)給核心帶正電同時消除表面負電荷;2)通過二硫鍵將小核酸藥物連接在樹枝狀聚合物。這些結構可以通過加入PEG進一步穩定。樹枝狀聚合物的模塊化允許進一步設計和調節載體的功能,提高藥物的靶向特異性和遞送效率。
多肽
聖諾製藥的多肽納米顆粒(PNP)技術。PNP是由人工設計和合成的組氨酸和賴氨酸的多肽共聚物。在水溶液,共聚物分子中的賴氨酸的氨基與核酸分子中的磷酸基團通過離子鍵相互作用而結合,自組裝形成一定大小的納米顆粒。藥物納米顆粒通過細胞的內吞作用進入細胞。在內含體內,由於酸性增強,促使弱鹼性的組氨酸開始質子化,進而導致內含體膜溶解,釋放內含的小幹擾核酸分子。
6.2.4 細胞外囊泡遞送系統
細胞外囊泡是未來最具潛力的遞送系統之一。細胞外囊泡(extracellular vesicles, EVs)作爲一種天然藥物遞送體系在近年來持續受到科研人員的關注。EVs是由細胞釋放的各種具有膜結構的囊泡的統稱,作爲細胞本身重要的通訊手段,EVs通過在鄰近細胞間運送核酸及蛋白質來協助完成細胞間的交流活動,因此與現有的載藥體系相比,細胞外囊泡天然具有能夠逃避吞噬作用、延長藥劑體內半衰期以及降低免疫原性的屬性。根據起源細胞不同、大小和組成異質性,EVs主要分爲外泌體(Exosomes)、微囊泡(Microvesicles)、凋亡小體(Apoptotic body)和腫瘤小泡(Large oncosomes),其中,適合作爲藥物遞送載體的主要是外泌體和微囊泡。
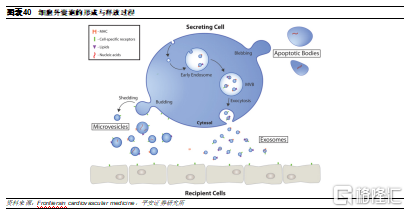
目前實現細胞外囊泡載藥的方法可以大致分間接載藥和直接載藥:
間接載藥是通過處理細胞外囊泡母細胞來實現細胞外囊泡載藥的,載藥效率較低;
直接載藥是將待載藥物直接裝入細胞外囊泡中,載藥效率更高。
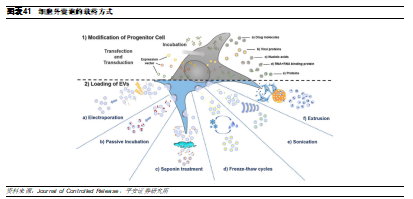
EVs的挑戰主要在於工藝開發方面。但由於EVs屬於生物製劑,因此其活性物質較爲複雜,面臨着一定程度的內在生物變異,可能導致不同批次間產品的異質性,對於開發工藝的挑戰難度較大。此外,EVs目前生產和發揮作用的機制也尚未明確,因此對其成藥性和體內分佈仍然有待進一步研究,目前仍然處於探索階段。未來如果技術難關被解決,EVs將有望爲小核酸藥物領域帶來突破。
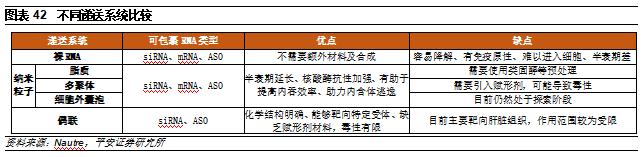
6.2.5 遞送系統比較
目前僅LNP與GalNAc兩種載體已有藥物獲批上市,因此在工藝成熟度和成藥性上相比其他載體技術平臺更爲成熟,目前其他平臺仍然處於驗證階段,存在失敗的可能性。
07
小核酸藥物主要技術平臺和領軍企業
7.1 Alnylam
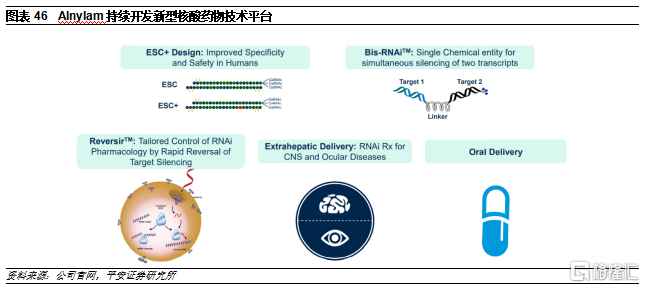
Alnylam是siRNA領域的龍頭企業。公司成立於2002年,2004年納斯達克上市,目前市值160億美元。公司上市以來市值已上漲超20倍。公司一直深耕RNAi治療領域,相繼開發了多個遞送平臺,包括第二代脂質納米粒子遞送平臺(DLin-MC3-DMA)和GalNac遞送平臺,均是RNAi藥物遞送領域的突破性技術。由於GalNac相比LNP遞送系統具有顯著優勢,因此除了已獲批的Patisiran使用了LNP,目前公司在研產品基本均使用GalNac偶聯遞送系統。爲了進一步提高遞送效率和安全性,公司也在持續更新迭代GalNac平臺,從STC到ESC再到ESC+,不斷完善小核酸藥物開發技術。
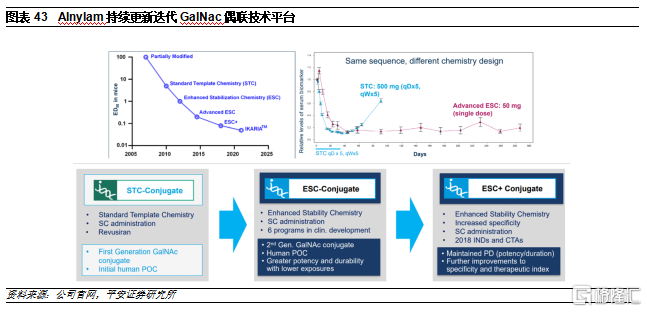
1)STC(Standard Template Chemistry):第一代GalNAc技術平臺,用於開發了Revusiran。
2)ESC(Enhanced Stability Chemistry):通過對siRNA片段進行化學修飾增強了藥物的穩定性以及肝臟的靶向性,其效力較第一代相比提高了10倍以上。但仍有可能在肝臟沉默其它非靶點基因,從而導致肝臟毒性。
3)ESC+:Alnylam的研發人員發現,RNAi脫靶效應的一個主要原因是與靶mRNA結合的siRNA種子區域部分序列可能具有類似miRNA的功能,可以與其它mRNA的3’非轉錄區結合,從而導致脫靶。在ESC+平臺中,研究人員在siRNA片段中添加了乙二醇核酸(GNA)。GNA具有熱不穩定性,因此若只結合種子區域中的部分序列無法發揮沉默作用,降低脫靶概率,進一步提高了RNAi療法的特異性和安全性。目前已開發出多款在研療法中已經得到應用並且獲得概念驗證。
4)IKARIA:超高效,可以做到>90%的沉默,同時一年只需要注射一次,有望能生產具有長效、可逆作用的小核酸藥物。目前已開發出藥物ALN-TTRsc04,但仍然處於臨牀前階段。
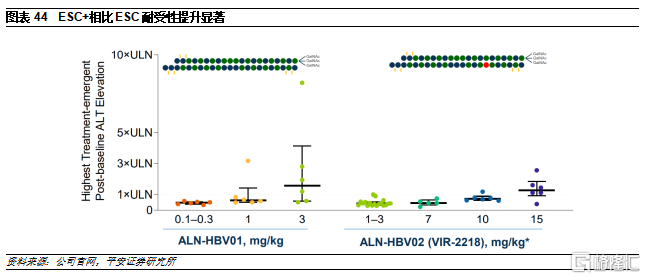
公司致力於開發出靶向其他組織的偶聯遞送系統。受益於豐富的研發經驗,公司在其他基於偶聯的siRNA遞送技術上也取得了持續的進展,將siRNA通過一個穩定、特異性的連接子與靶向不同組織或細胞的配體偶聯在一起,有望能夠提高RNAi療法在治療肝臟之外組織時的療效和穩定性。爲進一步拓展RNAi療法的應用範圍,Alnylam還與PeptiDream就肽-siRNA偶聯藥物開發達成合作,將共同選擇和優化肽,通過與靶細胞上表達的受體的特異性相互作用,將小幹擾RNA(siRNA)分子定向輸送到多種類型細胞和組織。
公司通過開發新型技術加寬護城河。還在開發多款新型小核酸藥物技術,包括雙靶點小核酸、可逆核酸藥物、核酸藥物口服劑型等。其中,雙靶點核酸藥物是將兩個siRNA通過可裂解linker連接在一起,並使用GalNac等偶聯技術靶向目標組織,從而可以持久穩定的同時沉默兩個目標基因,有望能夠進一步解決腫瘤異質性難題以及感染類疾病的耐藥性問題。
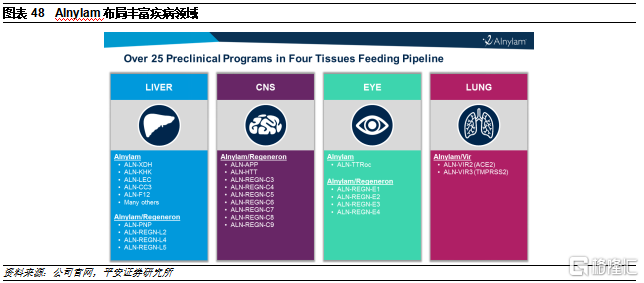
公司具有豐富研發管線。公司目前已有4款RNAi藥物完成上市,分別爲Onpattro(2018上市,用於治療遺傳性甲狀腺素介導的澱粉樣變性的多發性神經病)、Givlaari(2019上市,用於治療急性肝卟啉症)、Oxlumo(2020上市,用於治療原發性高草酸尿1型)和Leqvio(2020上市,用於降低低密度脂蛋白膽固醇。此外,還有4款產品已進入臨牀後期階段,將在未來5年內陸續完成上市,有望持續增厚公司業績表現。但從適應症來看,公司現階段的產品多是基於GalNac平臺開發,因此主要集中在肝臟相關疾病領域。

伴隨公司持續開發新的遞送系統,公司臨牀前研發管線中已佈局腦部、眼部和肺部領域疾病,持續鞏固公司的行業領軍地位,豐富產品管線佈局。
7.2 Ionis
Ionis是ASO領域的領軍企業。公司創立於1989年,是ASO藥物研究和開發的領頭羊。公司的核心技術平臺爲配體共軛反義技術(Ligand Conjugated Antisense,LICA),其原理是將配體與細胞表面受體特異性偶聯,從而將藥物遞送至目標細胞和組織,例如將GalNac偶聯至ASO藥物。除了LICA技術外,公司還有2個核心修飾技術,共同推動新一代ASO藥物開發:
第2代修飾:由於天然ASO在體內會被酶降解並且半衰期較短,因此公司開發了第2代ASO修飾,即MOE gapmer,採用了骨架PS修飾以及其呋喃核糖基環上的2’-氧-2-甲氧乙基(2’-MOE)取代修飾。通過二代修飾的藥物通常是一種gapmer設計 (5-10-5 gapmer):在寡核苷酸的5’端和3’端有5個2’-MOE修飾的核苷酸,中間有10個DNA核苷酸,以支持RNase H作用,例如首個靶向RNA的hATTR治療藥物Tegsedi(Inotersen)。
第2.5代修飾:是一種核苷酸橋接修飾,通過使用cEt糖基取代,使核苷酸的第2和第4個碳原子之間橋連,從而形成雙環核苷結構,進一步增強穩定性。這種取代相比於第2代修飾其效能提高了約10倍。cEt技術是公司目前最先進的反義技術之一,用於生產下一代ASO藥物。

公司擁有小核酸領域的首個爆款產品,研發管線豐富。基於研發平臺,公司建立了豐富的研發管線,目前已有三款ASO藥物獲批上市,分別爲Tegsedi(2018年上市,用於治療遺傳性轉甲狀腺素蛋白澱粉樣變性患者的多發性神經病)、Spinraza(2019年上市,用於治療脊髓性肌萎縮症)、waylivra(2019年上市,用於治療家族性乳糜微粒血癥綜合徵),其中,Spinraza在2019年的收入已經超過20億美元,成爲RNAi治療重磅藥物。此外,公司還有超過三十多個在研品種,適應症覆蓋心血管、代謝、神經、呼吸系統、眼科、癌症、傳染病等領域。

7.3聖諾製藥
聖諾製藥是目前國內發展較爲靠前的小核酸企業。公司成立於2007年,專注於以核酸幹擾(RNAi)技術爲核心的新藥開發,憑藉豐富的經驗和較強的研發能力,公司開發了多個遞送平臺,包括PNP、GalAhead和GalNAc-PDoV平臺等,並且均可以攜帶單個或多個siRNA,從而使多個靶基因沉默,進一步提高療效:
PNP遞送系統
是基於一種天然可降解的多肽分子,由支化組氨酸賴氨酸聚合物(HKP)組成,當將HKP以適當比例和RNA混合時,會自動組合成包裹RNA的納米顆粒,包封率高達97%。注射到體內後,藥物可以通過非網格蛋白的內吞作用或NRP1受體傳遞至靶組織和細胞中。在進入細胞後,組氨酸介導的質子化會導致內含體裂解,從而提升內含體逃逸效率,促進siRNA釋放到細胞質中的細胞作用位點。由於多肽及RNA的生物降解性,PNP具有很高的安全性。此外,PNP納米粒子可以一次捕獲並封裝多個RNA分子,可以用於開發多靶點核酸藥物,同時沉默具有協同效應的兩個不同靶基因,增強藥物效果。
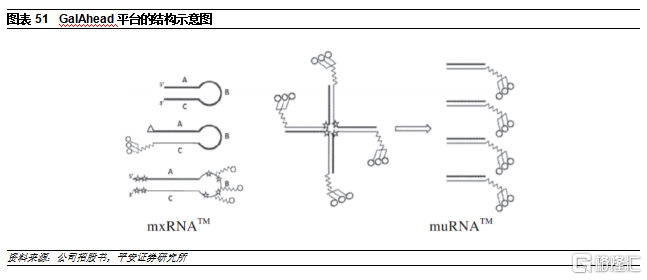
GalAhead
公司的GalAhead遞送平臺是強化版GalNac技術,可以用於下調單類基因(mxRNA,小型化RNAi觸發器),以及用於下調多類基因(muRNA,多單位RNAi觸發器)。
1)mxRNA:是最小的RNAi觸發器之一,由長度爲32個核苷酸的單鏈寡核苷酸組成,形成小發卡結構。GalNac部分可以共價結合在寡核苷酸的一個或多個位置,由於mxRNA技術每個RNAi觸發器只需合成一個寡核苷酸,而傳統GalNac偶聯藥物需要合成兩種寡核苷酸,因此mxRNA製造相對更加方便
2)muRNA:由多條長度約32個核苷酸的單鏈寡核苷酸組成,帶有共價連接的GalNac單糖。當混合時,寡核苷酸自組裝成多重結構,在進入細胞後,寡核苷酸顆粒可控地分解,產生多個單獨的RNAi觸發器,從而允許同時敲低多個目標,提高治療效率。
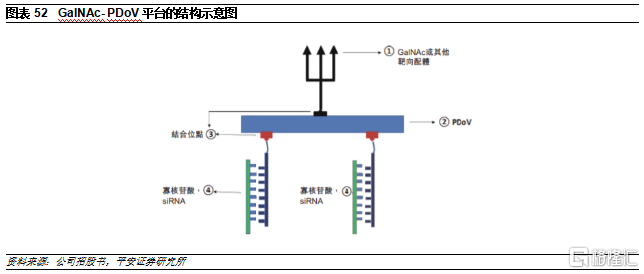
GalNAc肽對接載體(PDoV)遞送平臺
除了GalAhead,公司還開發了GalNAc-PDoV升級平臺。PDoV是一種由組氨酸-賴氨酸肽序列組成的遞送平臺,在一個位置用GalNac或其他靶向配體修飾,在其他位置用1-2個siRNA序列通過其正義鏈的骨架偶聯。在進入細胞後,與PNP遞送系統相似,PDoV部分中的組氨酸會在酸性環境中質子化,從而增強siRNA的內含體逃逸效率,因此與傳統GalNac技術相比其遞送效率更高。此外,GalNAc-PDoV平臺也可以同時遞送兩個siRNA,發揮協同作用。
基於豐富的遞送平臺技術,公司目前已經自主開發了十餘款核酸藥物,治療領域覆蓋腫瘤、纖維化、抗病毒、代謝、心血管疾病、肝、肺疾病。
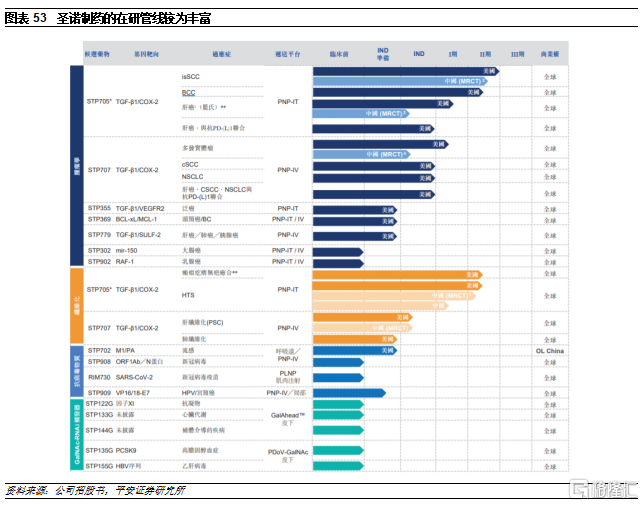
進展最快的STP705已經獲得積極II期數據。STP705是公司通過PNP將兩種靶向TGF-β1和COX-2 mRNA的siRNA組合而成,有望協同促進腫瘤抑制並下調參與纖維化作用的基因表達。STP705目前已開展多項臨牀試驗,包括膽管癌、肝細胞癌、非黑色素瘤皮膚癌和增生瘢痕治療等,其中,用於治療膽管癌、肝細胞癌和原發硬化性膽管炎的臨牀指症已經獲得了美國FDA的“孤兒藥”認證。STP705在成年患者中治療鱗狀細胞皮膚癌(isSCC)的2a期研究顯示了積極的療效和安全性結果,患者中有76%(19/25)在治療後病竈內腫瘤細胞組織學徹底清除。兩個最佳劑量組中達到了90%(9/10)的患者到達腫瘤細胞的組織學徹底清除。該研究沒有出現任何重大或嚴重的不良事件,包括沒有明顯的皮膚反應,公司得以在後階段研究中提前明確治療窗口。
7.4瑞博生物
瑞博生物逐步完善小核酸藥物全產業鏈佈局。公司成立於2007年,是國內最早成立的小核酸製藥企業之一,擁有豐富的小核酸藥物行業經驗和技術知識,建立了包括小核酸序列設計及高通量篩選、小核酸藥物遞送技術、小核酸穩定化修飾、小核酸藥物生物分析、小核酸藥學研究、小核酸單體研發等在內的自主可控、全技術鏈整合的六大核心技術平臺,可支持小核酸藥物從早期研發到產業化的全生命週期。
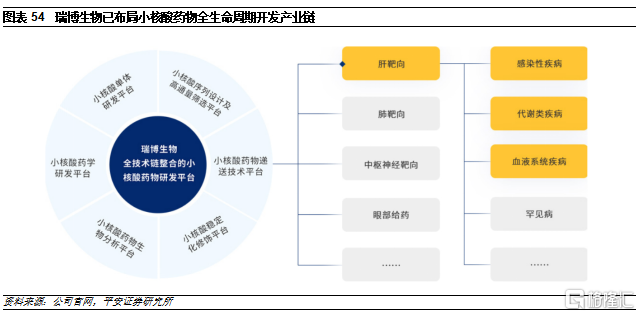
公司的技術平臺主要包括GalNAc和脂質體。公司使用了GalNAc肝靶向遞送技術,並圍繞肝源靶點開發了針對感染性疾病、代謝類疾病、血液系統疾病和罕見病等領域的諸多產品。同時,公司積極開發肺靶向、中樞神經靶向、局部給藥等遞送系統,以期實現其他器官組織的遞送突破,開展相關適應症的品種開發。此外,公司通過與LTC公司合作,掌握了基於脂質體的小核酸遞送技術,形成了在遞送技術上的技術儲備。憑藉技術平臺,公司目前已開展了十餘款產品的研究,主要圍繞糖尿病、腫瘤、眼科疾病、乙肝、高血脂等疾病領域開展,其中研發進度靠前的管線主要是通過合作引進的產品。
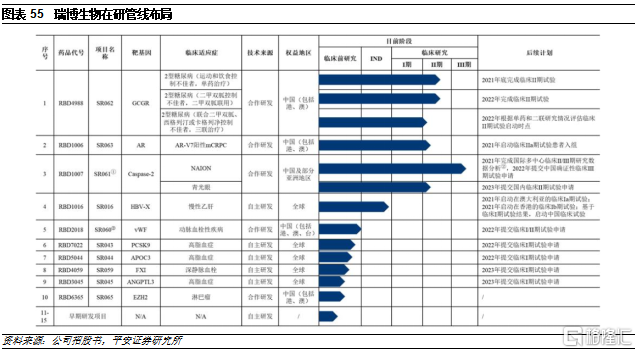
目前公司進度最快的產品是引進的SR061。該款藥物是公司從誇克合作引進的siRNA藥物,靶向半胱天冬酶2(Caspase 2),其首個開發的臨牀適應症爲非動脈炎性前部缺血性視神經病變(NAION),同時適用於包括青光眼在內諸多視神經損傷相關的眼科適應症。SR061是我國首款獲批臨牀試驗的小核酸藥物。NAION尚無確證有效的治療藥物,患者面臨着無藥可用的困境。SR061作爲全新機制的視神經保護劑,有望彌補未被滿足的臨牀需求。
08
投資建議
小核酸藥物行業靜待花開。小核酸藥物相比現有小分子和抗體藥物具有藥物靶點篩選快、研發成功率高、不易產生耐藥性、更廣治療領域和長效性等優點,並且有望從“源頭”解決疾病,具有較大發展潛力。從目前已有的藥物臨牀數據來看,小核酸藥物已經初步展現出治癒疾病、替代現有療法和填補空白適應症的潛力。目前小核酸藥物處境類似10年前的抗體藥物,仍然存在部分技術難關有待突破,包括肝臟以外組織的特異性遞送難題以及早期脫靶發現等問題。參考Alnylam從I期到III期接近60%的成功率,一旦技術問題實現突破,小核酸藥物有望迎來行業快速發展階段,成爲繼小分子和抗體藥物之後的第三代主流藥物,全面革新現有療法。
技術平臺是企業的護城河。從藥物的設計來看,相比抗體和小分子藥需要識別某些蛋白質複雜的空間構象,小核酸藥物的設計僅需靶標mRNA序列,因此早期候選藥物設計較爲快速,且不需要大規模藥物篩選,目前來看藥物序列設計壁壘相對較低。而主要的技術專利壁壘在於化學修飾和遞送系統,這也是小核酸藥物開發的核心。從目前行業內領軍企業的特點來看,這兩個關鍵技術具有平臺,可用於開發不同靶點的小核酸藥物,形成豐富的產品梯隊。因此,技術平臺是小核酸藥物開發企業的護城河,有望保障公司的持續造血能力,形成先發優勢,建議關注具有自主技術平臺的企業。
我國企業仍然有待實現專利創新突破。從全球小核酸藥物行業發展情況來看,我國起步相對國外較晚,目前LNP和GalNAc-siRNA綴合物技術的核心專利仍然掌握在國外企業手中,國內企業需要獲得相關授權或從零起步開發自主技術平臺,因此技術處於早期積累和認知階段。國內少數企業已經逐步開發出具有自主知識產權的遞送技術平臺,成爲國內小核酸藥物領域的開拓者。未來伴隨國內企業技術能力的進一步提升,我國小核酸行業將有望駛入發展快車道。
小核酸原料藥生產能力以及配套研發服務也是行業發展的關鍵。小核酸原料藥生產使用固相合成技術,其合成配套設備、潔淨環境等前期投入非常大,同時生產需符合GMP要求,因此在工藝開發、工藝放大和質量控制上存在較高壁壘,國內有能力生產小核酸原料藥的企業目前較爲稀缺。隨着市場需求的增加,未來小核酸藥物原料的及時供應成爲產品開發和商業化成功的重要挑戰。其中,核苷單體是小核酸原料藥的關鍵物料,目前全球僅有爲數不多的合格供應商。伴隨國內小核酸藥物進入研發爆發期,行業需求提升將驅動原料藥市場規模擴大,此外,具備相應研發生產服務能力CXO企業也將受益於行業發展,建議關注具有相關能力的企業。
09
風險提示
9.1 研發失敗風險
小核酸藥物目前多數仍然研發階段,新藥研發受到資金、政策、技術等多因素影響,存在進度不及預期甚至失敗的可能,此外,由於小核酸藥物開發仍然存在較多技術難關,未來如果無法突破可能會出現行業再次進入低谷期的情況。
9.2被新技術取代風險
小核酸藥物是作用於RNA的藥物,雖然相比蛋白藥物具有更顯著的優勢,但伴隨基因編輯等DNA技術的快速發展,有可能未來會出現被取代的情況。
9.3競爭加劇風險
未來可能會有更多企業佈局小核酸領域,導致競爭局面惡化。
9.4政策風險
作爲新興技術,行業監管政策存在發生變動的風險。


More Content